
The ASCO Post is pleased to present Hematology Expert Review, an ongoing feature that quizzes readers on issues in hematology. In this installment, Syed Ali Abutalib, MD, and L. Jeffrey Medeiros, MD, explore the key epidemiologic, pathologic, diagnostic, and prognostic aspects of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and monoclonal B-cell lymphocytosis (MBL). For each quiz question that follows, select the one best answer. The correct answers and accompanying discussions appear below.
GUEST EDITORS

Syed Ali Abutalib, MD

L. Jeffrey Medeiros, MD
Dr. Abutalib is Director of the Hematoloic Malignancies, Hematopoietic Stem Cell Transplantation & Cellular Therapy Programs at the Advocate/Aurora St. Luke’s Medical Center, Milwaukee, and Associate Professor at Rosalind Franklin University of Medicine and Science, Chicago. Dr. Medeiros is Professor and Chair, Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston.
Introduction
CLL/SLL and MBL form a continuum of mature B-cell lymphoproliferative disorders. CLL/SLL is diagnosed by examination of peripheral blood or by tissue biopsy, supported by characteristic immunophenotypic and molecular findings. Prognostic biomarkers, such as TP53 mutation and immunoglobulin heavy chain variable (IGHV) region sequencing, are essential for guiding patient management.
Designation as CLL requires a monotypic B-cell count in blood that is ≥ 5 × 109/L. The designation of SLL, by contrast, is based on tissue infiltration by a neoplasm in a patient with lymphadenopathy ≥ 1.5 cm and/or organomegaly in a patient with a peripheral blood B-cell count < 5 × 109/L.
MBL is a precursor lesion in which patients have monotypic B cells in the peripheral blood < 5 × 109/L without substantial lymphadenopathy (< 1.5 cm) or organomegaly. More than one type of MBL exists: (1) low-count CLL-like MBL, in which cells with CLL-like features are very few in the blood (< 0.5 × 10⁹/L); 2) high-count CLL-like MBL, in which cells with CLL-like features are numerous, but the overall monotypic B-cell count is < 5 × 109/L, and the patient does not have lymphadenopathy or organomegaly; and (3) non–CLL-like MBL, in which the cells do not have an immunophenotype like CLL, and the patient does not have lymphadenopathy or organomegaly. Many cases in the third group may be a precursor of marginal zone lymphoma.
Patients with CLL/SLL can transform into diffuse large B-cell lymphoma (DLBCL). In about 80% of patients, DLBCL is clonally related to CLL/SLL, and this is known as Richter transformation. In 20% of patients, DLBCL may be clonally unrelated. Comparative IGHV studies help to determine clonal relationships. The term accelerated phase of CLL/SLL is used to designate patients at higher risk of subsequently transforming to DLBCL. Criteria to support the designation of accelerated phase have been developed for tissue biopsy specimens and include the following: (1) large proliferation centers (> 20 × field); (2) > 2.4 mitoses/proliferation center; or (3) > 40% Ki67-positive cells in proliferation centers.
CLL also may undergo prolymphocytic progression with increased prolymphocytes in the blood. A minimum of 15% prolymphocytes is required to support the designation of prolymphocytic progression, which may be analogous to accelerated phase of CLL/SLL. When prolymphocytes are more numerous, at least 55% of cells in the blood smear, patients usually have an aggressive disease course. In the past, this finding was designated as B-cell prolymphocytic leukemia (no longer acceptable) or prolymphocytoid transformation of CLL/SLL.
Question 1
Which of the following criteria best distinguishes chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) from monoclonal B-cell lymphocytosis (MBL)?
A. Presence of prolymphocytes > 15% in peripheral blood
B. Detection of TP53 mutation or alteration
C. Absolute B-cell count ≥ 5 × 10⁹/L in the peripheral blood or presence of lymphadenopathy/organomegaly
D. Detection of CLL/SLL-phenotype B cells with a clonal count < 0.5 × 10⁹/L
E. Patients with MBL typically have bone marrow involvement of 30%; a primary diagnosis of CLL/SLL is not fulfilled unless peripheral blood B cells are ≥ 5 × 10⁹/L or there is lymph node enlargement (SLL).
Question 2
Which of the following statements is most accurate regarding the epidemiology and progression risk of CLL/SLL-type MBL?
A. CLL/SLL-type MBL is most prevalent in individuals younger than age 40 years and is rarely seen in elderly populations.
B. The risk of progression to CLL/SLL is highest among individuals with low-count MBL and is not influenced by the clonal B-cell count.
C. MBL is a clinically aggressive disorder with a high annual transformation rate and a short time to treatment in all patients.
D. The prevalence of CLL/SLL-type MBL increases with age and may reach up to 75% in individuals older than age 90.
E. Flow cytometry is not useful in detecting or characterizing CLL/SLL-type MBL due to its low sensitivity.
Question 3
Which of the following best explains the central mechanism driving the proliferation and survival of CLL/SLL cells in lymphoid tissues?
A. Constitutive activating mutations in the B-cell receptor (BCR) pathway
B. Autonomous T-cell signaling leading to B-cell clonal expansion
C. Antigen-driven BCR signaling supported by microenvironmental interactions
D. Overexpression of immunoglobulin genes due to chromosomal translocations
E. Loss of germinal center architecture and somatic hypermutation in all cases
Question 4
Which of the following is considered an essential diagnostic criterion for CLL/SLL?
A. Absolute B-cell count > 5 × 10⁹/L with classic CLL morphology and confirmatory flow cytometry findings
B. Detection of CD10 and SOX11 expression in a lymph node biopsy specimen
C. Presence of BTK or phospholipase C gamma 2 (PLCG2) mutations in peripheral blood
D. Complex karyotype and MUM1/IRF4 expression in proliferation centers
E. CD79b and FMC7 strongly positive with high CD20 expression
Question 5
Which of the following is most accurate regarding the molecular diagnostic workup for patients with CLL/SLL prior to initiating therapy?
A. TP53 mutation analysis is unnecessary if del(17p) is not detected by fluorescence in situ hybridization (FISH).
B. IGHV somatic hypermutation status must be retested during disease progression.
C. Translocations involving immunoglobulin gene loci are common in CLL/SLL and define prognosis.
D. BTK and PLCG2 mutations are typically present at diagnosis and guide initial treatment selection.
E. Comprehensive TP53 assessment should include both FISH for del(17p) and sequencing for TP53 mutations before therapy begins.
Question 6
Which of the following statements is most accurate regarding the assessment of IGHV somatic hypermutation status in patients with CLL/SLL?
A. IGHV somatic hypermutation status should be reassessed at every relapse due to its dynamic nature.
B. IGHV somatic hypermutation testing is recommended prior to treatment and needs to be performed only once per patient.
C. Patients with germline identity < 98% are considered IGHV-unmutated and have a worse prognosis.
D. IGHV somatic hypermutation testing is useful only for determining the presence of subset #2 configuration.
E. IGHV mutation status can be accurately inferred from standard flow cytometry panels.
Answers to Hematology Expert Review Questions
Question 1
Which of the following criteria best distinguishes chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) from monoclonal B-cell lymphocytosis (MBL)?
Correct Answer: C. Absolute B-cell count ≥ 5 × 10⁹/L in the peripheral blood or presence of lymphadenopathy/organomegaly.
Expert Perspective
The most important factor distinguishing CLL/SLL from MBL is the absolute clonal B-cell count in peripheral blood and the presence of clinical features, such as lymphadenopathy or organomegaly. A diagnosis of CLL/SLL is made when the B-cell count is ≥ 5 × 10⁹/L, or there is evidence of tissue involvement, such as enlarged lymph nodes, which would classify the condition as SLL. In contrast, MBL is defined by a B-cell count < 5 × 10⁹/L and the absence of these clinical features.
Although option 5 contains accurate information, including the note that bone marrow involvement alone does not establish a diagnosis of CLL/SLL, the most direct and defining criterion remains the peripheral blood B-cell count and/or lymphadenopathy, as stated in option 3. Options 1 and 2 relate to disease progression or prognosis rather than diagnosis, whereas option 4 refers to a specific MBL subtype (low-count MBL) but does not distinguish MBL from CLL/SLL as clearly as option 3.
Question 2
Which of the following statements is most accurate regarding the epidemiology and progression risk of CLL/SLL-type MBL?
Correct Answer: D. The prevalence of CLL/SLL-type MBL increases with age and may reach up to 75% in individuals older than age 90.
Expert Perspective
CLL/SLL-type MBL is an age-associated condition, with its prevalence increasing significantly in older populations. High-sensitivity flow cytometry can detect clonal B-cell expansions in 5% of individuals between the ages of 40 and 50 years, up to 25% in those between the ages of 65 and 80 years, and as high as 50% to 75% in individuals older than age 90. This makes option D the most accurate statement.
In contrast, option 1 is incorrect, as younger individuals have a much lower prevalence CLL/SLL-type MBL. Option 2 is also incorrect because the risk of progression to CLL/SLL is significantly influenced by the clonal B-cell count—with counts > 3 × 10⁹/L associated with higher progression risk, whereas low-count MBL (< 0.5 × 10⁹/L) has a negligible risk. Option 3 is inaccurate; although some patients with higher B-cell counts and certain biomarkers (eg, unmutated IGHV, elevated β2-microglobulin > 3.5 g/L) may progress faster, most individuals with MBL remain stable for years, and only about 0.5% to 2% progress per year. Option 5 is incorrect because flow cytometry is essential in detecting and immunophenotyping MBL, particularly in identifying the characteristic CLL/SLL phenotype.
Question 3
Which of the following best explains the central mechanism driving the proliferation and survival of CLL/SLL cells in lymphoid tissues?
Correct Answer: C. Antigen-driven BCR signaling supported by microenvironmental interactions.
Expert Perspective
The pathogenesis of CLL/SLL is multifactorial but is predominantly driven by antigen-stimulated BCR signaling within the lymph node microenvironment. Unlike some other lymphoid malignancies, CLL/SLL cells do not usually harbor activating mutations in BCR pathway components. Instead, their growth and survival rely on interactions with antigens and stromal support cells—notably T-helper cells, nurse-like macrophages, and mesenchymal stromal cells within proliferation centers. These interactions stimulate BCR signaling through normal physiologic mechanisms, including recognition of microbial or autoantigens and even homotypic BCR-BCR interactions, a phenomenon contributing to BCR stereotypy. These processes drive downstream signaling via kinases such as spleen tyrosine kinase, Bruton’s tyrosine kinase (BTK), and phosphoinositide 3-kinase (PI3K), with BTK inhibitors being particularly effective therapeutically.
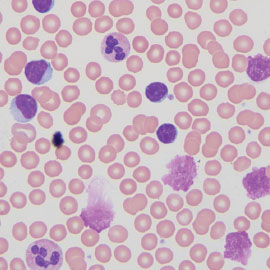
FIGURE 1: Blood smear of CLL showing many small lymphocytes and smudge cells (Wright-Giemsa, 1000×).
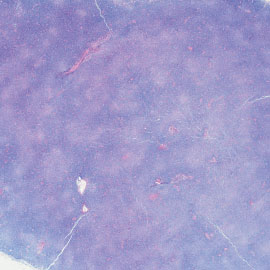
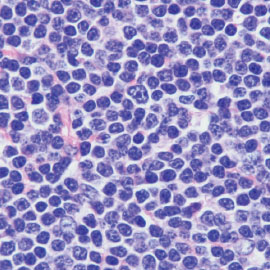
FIGURE 2: A. Low magnification showing replacement of lymph node by CLL/SLL. Note the many pale, nodular areas consistent with proliferation centers at this magnification. B. High magnification of CLL/SLL showing a mixture of small lymphocytes and scattered larger cells (prolymphocytes and paraimmunoblasts; hematoxylin-eosin, A. 20×; B. 1000×).
Option 1 is incorrect because activating mutations in the BCR pathway are not a common initiating event in untreated CLL/SLL. Option 2 misrepresents the role of T cells; although they are important in the microenvironment, they do not autonomously drive B-cell proliferation. Option 4 does not apply here, as chromosomal translocations involving immunoglobulin genes are more typical of other B-cell neoplasms, such as follicular lymphoma, mantle cell lymphoma, and Burkitt lymphoma. Option 5 is inaccurate since CLL/SLL includes both somatic hypermutation–positive (mutated) and somatic hypermutation–negative (unmutated) cases, which are linked to the cell of origin and clinical behavior rather than uniform absence of somatic hypermutation.
Question 4
Which of the following is considered an essential diagnostic criterion for CLL/SLL?
Correct Answer: A. Absolute B-cell count > 5 × 10⁹/L with classic CLL morphology and confirmatory flow cytometry findings.
Expert Perspective
The essential diagnostic criteria for CLL/SLL include a peripheral blood B-cell count > 5 × 10⁹/L, classic cytomorphologic features (small, mature-looking lymphocytes with scant cytoplasm and clumped chromatin), and a confirmatory immunophenotype by flow cytometry (Figures 1 and 2). This immunophenotype typically includes expression of CD5, CD19, CD20 (dim), CD23, and monotypic surface light chain (dim). Option 1 accurately captures these essential features and is thus the correct answer.
Option 2 is incorrect because CD10 and SOX11 are not expressed in CLL/SLL, and their presence would argue against the diagnosis, suggesting instead other types of lymphoma such as follicular lymphoma (CD10-positive) and mantle cell lymphoma (SOX11-positive). Option 3 involves BTK and PLCG2 mutations, which are relevant for prognosis and treatment resistance, especially in the context of BTK inhibitor therapy, but they are not required for diagnosis. Option 4 combines markers associated with proliferation centers (MUM1/IRF4) and poor prognosis (complex karyotype), but, again, these markers are desirable or supportive but not essential. Option 5 describes an immunophenotype more typical of non-CLL B-cell neoplasms (eg, mantle cell lymphoma), where FMC7 and CD79b are usually strongly expressed—this pattern is not typical for CLL/SLL.
Question 5
Which of the following is most accurate regarding the molecular diagnostic workup for patients with CLL/SLL prior to initiating therapy?
Correct Answer: E. Comprehensive TP53 assessment should include both FISH for del(17p) and sequencing for TP53mutations before therapy begins.
Expert Perspective
A thorough molecular workup is essential in CLL/SLL, particularly prior to initiating treatment, as it influences both prognosis and therapeutic decision-making. Among the most critical factors is the TP53 status, which requires dual assessment: FISH to detect del(17p) and sequencing to identify TP53 mutations. This testing is needed because up to 60% of patients with TP53 disruption have both the deletion and mutation, whereas around 30% may harbor TP53 mutations without del(17p). Hence, relying solely on FISH would miss a substantial number of patients with TP53 aberrations. TP53 alterations in CLL/SLL are associated with treatment resistance and poor prognosis, especially in the context of chemoimmunotherapy.
Option 1 is incorrect because TP53 mutation testing is necessary, even in the absence of del(17p). Option 2 is false; IGHV somatic hypermutation status is stable over time and does not need to be retested, as it reflects the cell of origin. Option 3 is misleading—immunoglobulin gene translocations are rare in CLL/SLL, with exceptions like IGH::BCL2 found in only about 2% of cases. Option 4 is incorrect because BTK and PLCG2 mutations typically arise during therapy, especially with BTK inhibitors, and are not present at diagnosis. Therefore, option 5 most accurately reflects current diagnostic guidelines.
Question 6
Which of the following statements is most accurate regarding the assessment of IGHV somatic hypermutation status in patients with CLL/SLL?
Correct Answer: B. IGHV somatic hypermutation testing is recommended prior to treatment and needs to be performed only once per patient.
Expert Perspective
IGHV somatic hypermutation status is an essential prognostic marker in CLL/SLL, guiding therapeutic decisions, especially in the era of targeted therapies. Testing for somatic hypermutation status of the clonotypic rearranged IGHV gene is recommended before the start of therapy, because it helps to stratify patients into mutated (better prognosis) and unmutated (poorer prognosis) categories. The distinction is based on the percentage of identity between the IGHV sequence in the patient’s leukemic clone and the closest germline gene. A germline identity below 98% defines IGHV-mutated CLL/SLL, whereas a germline identity ≥ 98% indicates IGHV-unmutated CLL/SLL. Since the rearranged immunoglobulin gene sequence is stable over time, this test only needs to be done once per patient.
Option 1 is incorrect because the IGHV status does not change over time and therefore does not require repeat testing. Option 3 reverses the definition—less than 98% indicates mutated, not unmutated status. Option 4 is misleading; although subset #2 configuration (IGHV3-21/IGLV3-21) is clinically important and mostly falls under the mutated group, somatic hypermutation testing serves a broader role beyond detecting this subset. Of note, subset #2 may exhibit aggressive clinical behavior despite being classified as mutated, underscoring the need for comprehensive IGHV analysis that goes beyond mutation status alone. Option 5 is incorrect because somatic hypermutation status cannot be determined by flow cytometry—it requires molecular sequencing of the IGHV gene.
DISCLOSURE: Dr. Abutalib has served on the advisory board of AstraZeneca. Dr. Medeiros reported no conflicts of interest.
SUGGESTED READINGS
- Khoury JD, Solary E, Abla O, eds: World Health Organization Classification of Haematolymphoid Tumours: Mature B-Cell Neoplasms: Pre-neoplastic and Neoplastic Small Lymphocytic Proliferations, 5th ed, p 912. Lyon, France; International Agency for Research on Cancer; 2024.
- Khoury JD, Solary E, Abla O, eds: World Health Organization Classification of Haematolymphoid Tumours: Monoclonal B-Cell Lymphocytosis, 5th ed, pp 113-120. Lyon, France; International Agency for Research on Cancer; 2024.
- Khoury JD, Solary E, Abla O, eds: World Health Organization Classification of Haematolymphoid Tumours: Chronic Lymphocytic Leukaemia/Small Lymphocytic Lymphoma, 5th ed, pp 121-161. Lyon, France; International Agency for Research on Cancer; 2024.