
GUEST EDITORS

Syed Ali Abutalib, MD

Amer M. Zeidan, MBBS, MHS
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic stem cell disorders that are characterized by ineffective hematopoiesis, resulting in cytopenias, and they carry a risk of progression to acute myeloid leukemia (AML). In 2022, the fifth edition of the World Health Organization (WHO) classification introduced the new term, myelodysplastic neoplasms (which will continue to be abbreviated as MDS), as it better delineates the neoplastic nature of MDS.1
The current landscape for the management of patients with lower-risk MDS is focused on the improvement of blood cell counts, decreasing transfusion burden, reducing complications of the disease, and thereby improving patients’ quality of life. None of the treatment options for patients with lower-risk MDS have been shown to improve overall survival, and this has not been a primary endpoint in clinical trials for lower-risk MDS. Platelet transfusions and occasional prophylactic antimicrobial therapy are also used for patients with significant thrombocytopenia or severe/profound neutropenia, respectively.2
IMerge Study
Abstract: Results from a phase III, randomized, double-blind, placebo-controlled study of imetelstat in patients with heavily transfusion-dependent non-del(5q) lower-risk myelodysplastic syndromes relapsed, refractory or ineligible to erythropoiesis-stimulating agents (ESAs; ClinicalTrials.gov identifier NCT02598661).3
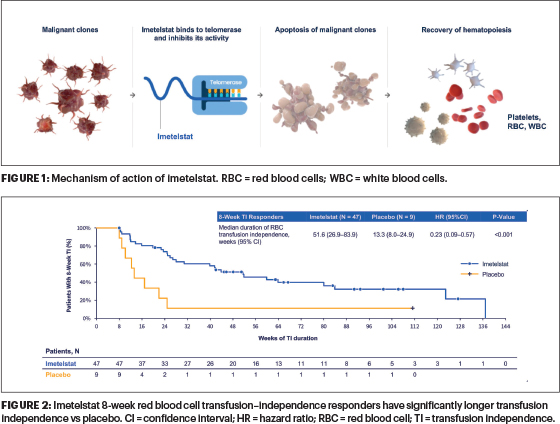
Background: The most common cytopenia experienced by patients with lower-risk MDS is anemia requiring frequent red blood cell transfusions until a therapeutic option is effective in eliminating this transfusion burden and its complications. Imetelstat is a first-in-class direct and competitive inhibitor of telomerase activity. Imetelstat specifically targets malignant clones in MDS with abnormally high telomerase activity and inhibits that activity, thereby enabling recovery of effective hematopoiesis (Figure 1).
In the second phase of the IMerge study, patients with lower-risk MDS who were heavily red blood cell transfusion–dependent, ESA–relapsed/refractory or ineligible, non-del(5q), and naive to lenalidomide and hypomethylating agents achieved durable and continuous transfusion independence when treated with imetelstat. Specifically, the 8-week transfusion-independent rate was 42%, with a median transfusion-independent duration of 86 weeks.4 This analysis reports phase III results from IMerge in the same patient population.
Methods: Heavily red blood cell transfusion–dependent patients with ESA-relapsed or refractory non-del(5q) lower-risk MDS naive to lenalidomide and hypomethylating agents were randomly assigned 2:1 to receive an intravenous infusion of imetelstat at 7.5 mg/kg or placebo every 4 weeks. The primary endpoint was 8-week red blood cell transfusion independence.
Results: Robust transfusion-independence rates were noted with imetelstat. The 8-week transfusion-independence rate was 39.8% with imetelstat vs 15.0% with placebo. Median transfusion-independence duration was significantly longer for imetelstat vs placebo, 51.6 vs 13.3 weeks (P < .001; Figure 2). Patients receiving imetelstat had significantly higher mean hemoglobin levels (P < .001) and fewer transfusions (P = .042) over time than those on placebo. The rate of 8-week transfusion independence was significantly higher with imetelstat vs placebo across subgroups: ring sideroblasts status, red blood cell transfusion burden status, International Prognostic Scoring System (IPSS) risk category, or serum erythropoietin status.
Regarding safety, severe clinical consequences from grade 3 or 4 neutropenia, febrile neutropenia, severe infection, thrombocytopenia, or bleeding were similar in patients treated with imetelstat and placebo. Further, more than 80% of the severe (grade 3 or 4) cases of neutropenia and thrombocytopenia associated with imetelstat use were reversible to grade ≤ 2 within 4 weeks with dose interruption or reduction.
With imetelstat, a greater reduction in SF3B1 variant allele frequency correlated with a greater increase in hemoglobin and longer red blood cell transfusion–independence duration. A greater reduction in variant allele frequency in TET2, DNMT3A, or ASXL1 also correlated with longer red blood cell transfusion–independence duration.
Clinical Implications: Imetelstat demonstrated highly statistically significant and clinically meaningful benefit compared with placebo in a heavily transfusion-dependent lower-risk MDS population. Safety results are consistent with prior imetelstat clinical experience with no new safety signals. Reduction in variant allele frequency of genes that are commonly mutated in MDS—SF3B1, TET2, DNMT3A, or ASXL1—and their correlation with clinical endpoints supports the disease-modifying potential of imetelstat. The U.S. Food and Drug Administration (FDA) has accepted a new drug application for imetelstat as a treatment for patients with transfusion-dependent anemia in lower-risk MDS, with a decision regarding approval expected by early 2024.
COMMANDS Study
Abstract: Efficacy and safety of luspatercept vs epoetin alfa in ESA-naive, transfusion-dependent, lower-risk MDS: Interim analysis of a phase III, open-label, randomized controlled trial (NCT03682536).5
Background: ESAs are the standard-of-care treatment for anemia in most patients with lower-risk MDS and an endogenous serum erythropoietin level < 500 U/L, but responses are limited and transient. Luspatercept-aamt is a recombinant fusion protein that binds transforming growth factor–beta superfamily ligands to reduce SMAD2 and SMAD3 signaling, allowing better red blood cell maturation by late-stage erythroblast differentiation. Luspatercept promotes late-stage erythroid maturation, as shown in the MEDALIST trial.6
Patients with lower-risk MDS without ring sideroblasts and with low transfusion burden may benefit from luspatercept therapy,7,8 which may result in increased mean platelet and neutrophil counts in patients with lower-risk MDS.8,9 Based on findings in the MEDALIST trial, luspatercept was originally indicated for the treatment of anemia, after failure of ESA treatment or for those who are unlikely to respond to ESAs, in adults with lower-risk MDS (per the revised IPSS [IPSS-R]) with ring sideroblasts or a myelodysplastic or myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, in cases requiring at least 2 red blood cell units per 8 weeks.
Methods: Eligible patients were aged 18 years or older, had a diagnosis of MDS of very low risk, low risk, or intermediate risk (per the IPSS-R), were ESA-naive with endogenous serum erythropoietin levels < 500 U/L, and required red blood cell transfusions (2 to 6 red blood cell units per 8 weeks for ≥ 8 weeks immediately before randomization). The primary endpoint was red blood cell transfusion independence for at least 12 weeks with a concurrent mean hemoglobin increase of at least 1.5 g/dL (weeks 1–24), assessed in the intention-to-treat population. Safety was assessed in patients who received at least one dose of study treatment.
Results: Luspatercept showed superiority vs ESAs, with approximately twice the number of patients achieving both transfusion independence and hemoglobin increase (common risk difference on response rate = 26.6, 95% confidence interval [CI] = 15.8–37.4, P < .0001; odds ratio = 3.1, 95% CI = 1.9–5.0). In addition, luspatercept delivered more durable responses, with nearly 2.5 years of median transfusion independence, which was approximately 1 year longer than ESAs.
The risk difference estimates for commonly mutated genes indicated that patients with mutations in ASXL1, TET2, SF3B1, and SF3B1 (defined as SF3B1 mutations with concomitant mutation of DNMT3A or ASXL1 and/or TET2) were more likely to have a primary endpoint response with luspatercept than with epoetin alfa.
Luspatercept has a manageable and predictable safety profile, consistent with previous clinical experience, and a convenient, every-3-week administration schedule.
Clinical Implications: Luspatercept is the first and only therapy to demonstrate superiority in a head-to-head study against ESAs and brings a paradigm shift in the treatment of lower-risk MDS–associated anemia. Luspatercept treatment also showed greater efficacy than epoetin alfa in SF3B1-mutated and ring sideroblast–positive subgroups of patients, as well as favorability in nonmutated SF3B1 and ring sideroblast–negative subgroups and across various somatic mutations associated with lower-risk MDS.
Nevertheless, further testing of long-term data and assessments of other subgroups of patients with lower-risk MDS will be needed to refine and validate the current findings. Based on the results of the COMMANDS trial, on August 28, 2023, the FDA expanded the approved luspatercept indication to include treatment of anemia in adults with very low– to intermediate-risk MDS who may require regular red blood cell transfusions and who have not received previous treatment with an ESA.
EQOL-MDS Study
Abstract: Eltrombopag for the treatment of lower-risk MDS with thrombocytopenia: Interim results of a phase II, randomized, placebo-controlled clinical trial (EQOL-MDS; NCT02912208).10
Background: In MDS, severe thrombocytopenia is associated with a poor prognosis. Treatment options for lower-risk MDS–associated severe thrombocytopenia are limited, especially in the presence of bleeding, aside from chronic platelet transfusions and hypomethylating agents. This multicenter trial presents the second-part long-term efficacy and safety results of eltrombopag in patients with lower-risk MDS and severe thrombocytopenia.
Eltrombopag is an orally bioavailable agonist of the thrombopoietin receptor. In adults with chronic immune thrombocytopenic purpura, eltrombopag rapidly increased platelet counts and significantly reduced bleeding episodes during treatment. In 2008, the FDA approved eltrombopag for the treatment of refractory or resistant immune thrombocytopenic purpura. Eltrombopag induced an increase in the megakaryocytic differentiation and in the formation of normal megakaryocytic colonies. These results provided the rationale for pursuing further research on eltrombopag for the treatment of thrombocytopenia in cases of MDS.
Methods: The phase II EQOL-MDS trial is a randomized, placebo-controlled, single-blind study (ie, participants do not know if they are receiving the placebo or the real treatment, but the researchers do) of adults with IPSS low- or intermediate-1–risk MDS. Patients with a stable platelet count (< 30 × 103/mm3) received eltrombopag (n = 112; starting dose of 50 mg once daily to maximum of 300 mg) or placebo (n = 57) until disease progression. Primary endpoints were duration of platelet response (calculated from the time of response to date of loss of response, defined as bleeding/platelet count < 30 × 103/mm3 or last date in observation) and long-term safety and tolerability.
Results: With 25-week follow-up (interquartile range = 14–68 weeks), platelet response occurred in 42.3% of eltrombopag recipients vs 11.1% in placebo recipients (odds ratio = 5.9, 95% CI = 2.3–14.9, P < .001). Among patients in the eltrombopag group, 25.5% lost the platelet response, with a cumulative thrombocytopenia relapse–free survival at 60 months of 63.6% (95% CI = 46.0–81.2 months).
Clinically significant bleeding (WHO bleeding score ≥ 2) occurred less frequently in the eltrombopag arm than in the placebo group (incidence rate ratio = 0.54; 95% CI = 0.38–0.75; P = .0002). Although no difference in the frequency of grade 1 or 2 adverse events was observed, a higher proportion of eltrombopag recipients experienced grade 3 or 4 adverse events (P = .002).
There was no difference in AML transformation between the two groups (9% vs 7%, P = .729).
Clinical Implications: Eltrombopag was effective and relatively safe in patients with lower-risk MDS with stable and severe thrombocytopenia. Higher platelet responses (42.3% vs 11.1%, P < .001) and decreased bleeding events (19.8% vs 31.5%, P = .0002) were observed in the eltrombopag arm compared with the placebo arm. n
DISCLOSURE: Dr. Abutalib has reported a financial relationship with AstraZeneca. Dr. Zeidan has served on advisory boards or consulted for and/or received honoraria from AbbVie, Agios, Pfizer, Astellas, Astex, ALX Oncology, Amgen, Akeso, BeiGene, BMS/Celgene, Boehringer Ingelheim, BioCryst, Chiesi, Daiichi Sankyo, Epizyme, Faron, Geron, Gilead Sciences, Genentech, Hikma, Ionis, Janssen, Kura, Keros, Karyopharm, Lava Therapeutics, Mendus, Notable, Novartis, Otsuka, Orum, Taiho, Takeda, Syndax, Sumitomo, STCube, Schrodinger, Servier, Syros, Vincerx, Regeneron, and Zentalis.
REFERENCES
1. Khoury JD, Solary E, Abla O, et al: The 5th edition of the World Health Organization classification of haematolymphoid tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 36:1703-1719, 2022.
2. Madanat YF, Xie Z, Zeidan AM: Advances in myelodysplastic syndromes: Promising novel agents and combination strategies. Expert Rev Hematol 16:51-63, 2023.
3. Zeidan AM, Platzbecker U, Santini V, et al: IMerge: Results from a phase 3, randomized, double-blind, placebo-controlled study of imetelstat in patients with heavily transfusion-dependent non-del(5q) lower-risk myelodysplastic syndromes relapsed/refractory to erythropoiesis stimulating agents. 2023 ASCO Annual Meeting. Abstract 7004.
4. Steensma DP, Fenaux P, Van Eygen K, et al: Imetelstat achieves meaningful and durable transfusion independence in high transfusion-burden patients with lower-risk myelodysplastic syndromes in a phase II study. J Clin Oncol 39:48-56, 2021.
5. Platzbecker U, Della Porta MG, Santini V, et al: Efficacy and safety of luspatercept versus epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): Interim analysis of a phase 3, open-label, randomised controlled trial. Lancet 402:373-385, 2023.
6. Fenaux P, Platzbecker U, Mufti GJ, et al: Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med 382:140-151, 2020.
7. Platzbecker U, Germing U, Götze KS,et al: Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol 18:1338-1347, 2017.
8. Platzbecker U, Götze KS, Kiewe P, et al: Long-term efficacy and safety of luspatercept for anemia treatment in patients with lower-risk myelodysplastic syndromes: The phase II PACE-MDS study. J Clin Oncol 240:3800-3807, 2022.
9. Garcia-Manero G, Mufti GJ, Fenaux P, et al: Neutrophil and platelet increases with luspatercept in lower-risk MDS: Secondary endpoints from the MEDALIST trial. Blood 139:624-629, 2022.
10. Oliva EN, Riva M, Niscola P, et al: Eltrombopag for low-risk myelodysplastic syndromes with thrombocytopenia: Interim results of a phase II, randomized, placebo-controlled clinical trial (EQOL-MDS). J Clin Oncol 9:JCO2202699, 2023.