

We are unlikely to manage this disorder with one or even several drugs in the way tyrosine kinase inhibitors have cured some people with CML. Different people are likely to need different therapies or combinations of therapies, which will need to be proved effective in controlled clinical trials.— Robert Peter Gale, MD, PhD, DSc(hc), FACP, FRSM
Tweet this quote
November is National Pancreatic Cancer Awareness Month, the impetus for this article. Pancreatic cancer is a huge health challenge. It's the eighth most common cancer in the United States and the fourth most common cause of cancer deaths but is expected to become the second most common cause of cancer deaths within a decade. It’s a disease whose biology we need to better understand, in hopes we can develop more effective therapies and perhaps ways to prevent it and/or to diagnose it earlier.
Here I consider several areas of recent interest, progress, and controversies including pancreatic cancer biology, mutational landscape, the concept of latency, and causes of pancreatic cancer. Detailed references are available at the end of this commentary.
Biology of Pancreatic Cancer
The biology of pancreatic cancer in animals and humans is discussed extensively in several recent reviews.1-3 The disease is the eighth most common cancer in the United States, with about 45,000 cases annually, and is the fourth leading cause of cancer death, with a 5-year survival rate < 10%.4-7 Median age at diagnosis is 65 to 75 years.
About 95% of pancreatic cancers are adenocarcinomas arising in the exocrine pancreas. The remaining 5% arise in the endocrine (islet) pancreas or supporting tissues. Adenocarcinomas are thought to arise in cells in the pancreas ducts. However, considerable data indicate plasticity of pancreas cells.8 Namely, acinar and/or centroacinar cells can differentiate into duct cells and/or migrate into the pancreatic ducts as part of the normal process of pancreatic cell replacement and maturation.8 The plasticity of pancreatic cells is illustrated in Fig. 1.8
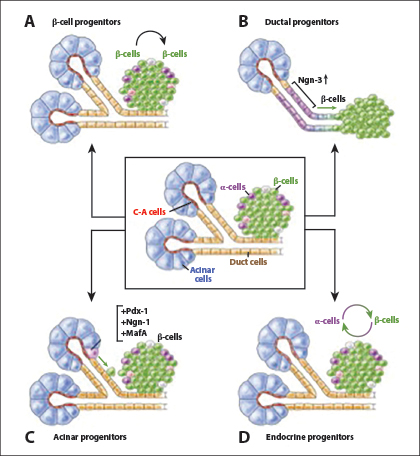
Fig. 1: Plasticity of Pancreatic Cells. Depiction of the structural elements of the exocrine pancreas including acinar, centro-acinar (C-A) and duct cells, and the endocrine islet including α- and β-cells. (A) Preexisting β-cells serve as progenitors for β-cells and expand via proliferation. (B) Upon injury, duct or duct-associated cells begin to express Ngn3, which could result in the formation of endocrine cells, including β-cells. (C) Viral expression of Pdx-1, Ngn-3, and MafA in adult acinar cells promotes reprogramming toward β-cells. (D) α-cells can transdifferentiate into β-cells following their depletion, whereas β-cells can become α-like cells or undifferentiated cells during stress or following deletion of Foxo1. Reprinted, with permission, from Stanger BZ, Hebrok M.8
Changes in the appearance of pancreas duct cells (whose origin may be in acinar cells as described previously) as they move from normal to neoplasm are well described and widely accepted.9,10 The process involves the progressive accumulation of somatic mutations. Acute and chronic inflammation—both clinical and subclinical—may accelerate this process by increasing cell proliferation.11
Other hypotheses for the development of pancreatic cancer include a role for supporting (stellate) cells, immune cells, and microenvironmental (stromal) abnormalities. However, these data are controversial. For example, pancreatic cancer incidence is not increased in persons with immune deficiency, including an inherited disorder of decreased immunity or acquired immune deficiency (such as acquired immune deficiency syndrome [AIDS] or disorders seen in persons receiving intensive immune suppression after organ transplants).
Studies of persons with pancreatic cancer receiving immune interventions, such as anti–programmed cell death protein 1 (PD-1), anti–programmed cell death ligand 1 (PD-L1) or anti–cytotoxic T-lymphocyte–associated protein-4 (CTLA-4) antibodies, other monoclonal antibodies, oncolytic viruses, vaccines, and adoptive cell therapy, are in progress. A ClinicalTrials.gov PUBMED search using the Boolean operators immune therapy AND pancreatic cancer identified 181 studies, 60 of which are recruiting subjects and 59 of which are completed.
Most of these trials are phase I studies of diverse advanced cancers with few subjects with pancreatic cancer. Six studies have reported results, some of which seemed favorable. However, results of one phase II study (ECLIPSE trial) and a second phase III study (IMPRESS trial) of immune therapy are disappointing. There may be subsets of persons with pancreatic cancer more likely to respond to immune checkpoint inhibitors such as those with microsatellite instability, a feature of pancreatic cancers with defective DNA repair protein functions. These cases may account for up to 20% of pancreatic cancers. Because of potential selection biases, data from phase III studies are needed to critically evaluate efficacy.
Mutation Landscape of Pancreatic Cancer
The mutation landscape of pancreatic cancer is reviewed in several reports of whole-exome and whole-genome sequencing of relatively large numbers of cases.1,11-15 Recently Bailey and coworkers used whole-genome and deep-exome sequencing, gene copy number analyses, and RNA expression profiles to study more than 400 cases of pancreatic cancer.15 Their report is data-dense, but some highlights follow.
Pancreatic cancer in humans is a progressive, multistep process probably occurring over a prolonged interval in most instances.— Robert Peter Gale, MD, PhD, DSc(hc), FACP, FRSM
Tweet this quote
Presumed driver mutations (which are fundamental to the disease process, in contrast with passenger mutations, which are not) were identified in over 20 significantly mutated genes. Mutations in KRAS, TP53, SMAD4, and CDKN2A were the most common, consistent with prior studies.14 These mutations affected diverse intracellular pathways and processes including activating KRAS mutations, DNA-damage repair, cell-cycle regulation (TP53, CDKN2A), TGF-ß signaling (SMAD4, SMAD3), histone modification, the SW1/SNF complex, the BRCA pathway (BRCA1, BRCA2, ATM, PALB2), WNT signaling, and RNA processing (SF3B1, U2AF1).
Interestingly, about one-third of the BRCA pathway–related mutations were germline and two-thirds were somatic. Some mutations such as those in SF3B1 were aggregated at specific hotspots (K700E), and some are potentially targetable. Other mutations such as those in DNA-damage repair mechanisms are likely to respond to platinum drugs.
Pathways identified in this study are consistent with prior reports of abnormalities in DNA-damage repair, cell-cycle regulation, TGF-ß signaling, chromatin regulation, and axonal guidance.12-14,16 However, the dominant mutational pathways were DNA deamination, ectopic APOBEC activity, BRCA deficiency, and DNA-mismatch repair.
Bailey and coworkers then used RNA-sequencing analyses and array-based studies of transcriptional pathways to divide pancreatic cancers into four subtypes termed squamous, aberrantly differentiated endocrine exocrine (ADEX), pancreatic precursor, and immunogenic, based on transcriptional networks. Three of the subtypes of this transcriptomic classification (not the immunogenic) are mostly concordant with morphologic classification of pancreatic adenocarcinomas in the Collisson system.17,18 Selected data from this study are shown in Fig. 2, which illustrates the molecular classes and transcriptional networks of pancreas ductal adenocarcinoma.
The major message of this analysis is that structural variation is a prominent mechanism of genomic damage in pancreatic cancer. Commonly mutated genes include KRAS, TP53, SMAD4, CDKN2A, and ARID1A. Many other genes are mutated at substantially lower frequencies. The median number of mutated genes is about 60 per cancer.
These data support the concept that pancreatic cancer in humans is a progressive, multistep process probably occurring over a prolonged interval in most instances. Compare this complexity to cancers where so-called targeted therapy is effective, such as chronic myeloid leukemia (CML), caused by one mutation, and acute myeloid leukemia (AML), with a median of five mutations.
Another interesting difference is the lack of fusion genes—common in hematologic cancers—in pancreatic cancer. Obviously, we are dealing with a different situation in pancreatic cancer, where developing targeted therapy will likely be substantially more challenging.19 Another issue we face is the complex interaction between the microenvironment and pancreas cancer cells. In some patients, pancreatic cancer, fibrous, or connective tissue (desmoplasia) comprises > 50% or even 90% of cancer.
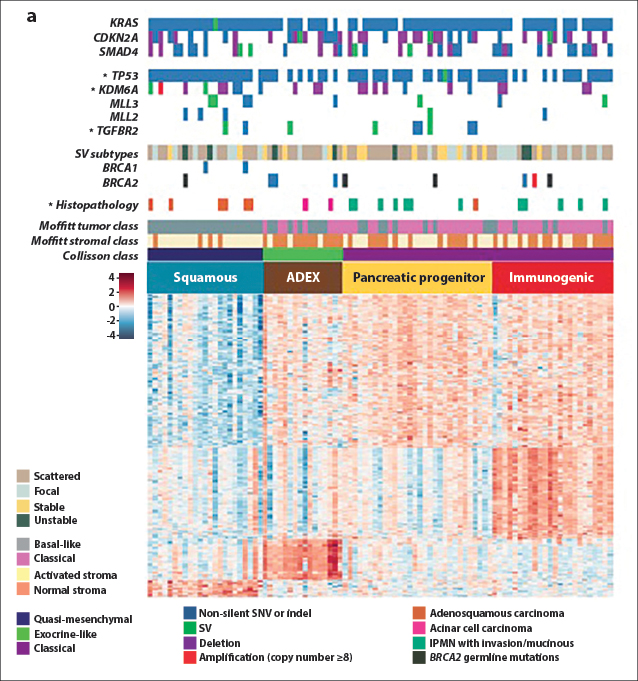
Fig. 2: Molecular Classes and Transcriptional Networks of Pancreatic Ductal Adenocarcinoma. Reprinted, with permission, from Bailey P, et al.15
How do these mutation landscape data help the clinician faced with treating pancreatic cancer? There are potential implications. For example, persons with DNA-repair abnormalities might be more likely to respond to platinum-based regimens. A question that arises is, should cancers in the immunogenic category be preferentially treated with the immune interventions discussed here. I urge caution: Supportive data are valuable, but statistics deal with associations, not cause and effect. And there is the challenge of identifying prognostic vs predictive biomarkers, a subtle but important distinction.
Finally, we have the KRAS mutation problem.1 KRAS is mutated in over 90% of pancreatic cancers in humans, and transgenic mice with mutated KRAS develop pancreatic cancer. Thus, although mutated KRAS seems a likely therapy target, it is presently not druggable. There are encouraging experimental data using small interfering RNAs, but they are presently far from clinically applicable.
Moreover, the genetic background of pancreatic cancer is far more complex than these data suggest. For example, although KRAS mutations are likely important in the development of pancreatic cancers, not all pancreatic cancers remain KRAS-dependent. Also, mutations in KRAS occur in older persons who never develop pancreatic cancer. This situation is analogous to the detection of mutations in AML-related genes such as DNMT3A, TET2, and ASXL1 in older persons—including some with clonal hematopoiesis—very few of whom develop a hematologic disorder during their remaining lifetime.20,21
Nevertheless, therapy directed against activated KRAS with small interfering RNAs is effective in some mouse models and is being tested in humans, as are downstream targets in the KRAS signaling pathway such as RAF, MEK, and ERK.
Pancreatic Cancer Latency
When does pancreatic cancer start, and how long is the interval between the first driver mutation, often in KRAS, and the clinical diagnosis? As discussed, the mutation landscape of pancreatic cancer is complex and suggests a need to reconsider the concept of cancer latency in general and in pancreatic cancer specifically. For example, compare CML caused by one fusion gene (BCR-ABL1) with pancreatic cancer linked to a median of 60 mutations and with no fusion genes.
This raises the question of precisely what we mean by latency of a cancer in humans. Is it the interval from the first driver mutation, like KRAS, or the last driver mutation to the diagnosis of a cancer or something else? And how do we account for the variability introduced by the increasing frequency of incidental cancer diagnoses, often from an unrelated radiologic procedure or blood testing. Some pancreatic cancers are diagnosed incidentally (eg, by a computed tomography scan for an unrelated reason) or are diagnosed years later than they might have been had there been active surveillance. Also, many cancers continue evolving genetically after their diagnosis, with unfolding transformations that might never end.
Another confounder (as discussed earlier) is that many mutations detected in pancreatic cancers are present in the pancreas of older persons who never develop pancreatic cancer in their lifetime. This situation is analogous to AML, where a substantial proportion of normal older persons have mutations in genes similar to those found in persons with AML.20,21 However, even when these seemingly normal people have clonal hematopoiesis, very few develop AML or any hematologic disorder in their lifetime.20,21
How can we know when a cancer in humans starts? One approach is to track the interval from radiation exposure to diagnosis of cancer in persons exposed to the atomic bomb explosions in Japan who later developed pancreatic cancer.. Although these data are controversial and the risk of developing pancreatic cancer is nonlinear, estimated latency varies over several decades. Admittedly, these data inform us of the latency of radiation-induced pancreatic cancer, which may differ from the latency of seemingly spontaneous pancreatic cancers.
A second way to estimate the lower boundary of latency is to consider the youngest age at diagnosis of pancreatic cancer. Although rare, pancreatic cancer (not pancreatoblastoma), typically of the acinar type, has been diagnosed in children as young as 1.5 years.22 This brief latency, clearly atypical, suggests some pancreatic cancers can develop rapidly. Perhaps this is so in persons with precancerous lesions such as those associated with diabetes or chronic pancreatitis (the former is controversial).
The relationship between diabetes and pancreatic cancer is well established but is also complex.— Robert Peter Gale, MD, PhD, DSc(hc), FACP, FRSM
Tweet this quote
Consider again CML, a much simpler cancer where we have a better chance to estimate latency. As discussed, CML is caused by one driver mutation (BCR-ABL1), necessary and sufficient to cause the disease. Using the A-bomb data, it seems CML can develop as quickly as 2 years after exposure or as late as 30 years or more thereafter.23,24 What accounts for this extraordinary variability in latency? My guess is that stochastic processes are responsible, but other hypotheses are also possible and much favored by nonstatisticians.
Another issue to consider is the 5% to 10% of persons with pancreatic cancer with mutations in DNA-repair genes such as BRCA1, BRCA2, and PALB2.25,26 Because proliferating cells replicate their DNA more often than nonproliferating cells and because mutations unavoidably accompany each round of DNA replication, persons with mutations in DNA-repair genes are likely to acquire additional mutations rapidly and develop cancer more rapidly than persons without these mutations.
Finally, different mutations confer different rates of cell proliferation and/or cell death (apoptosis) to normal or preneoplastic cells. Some mutations confer little or no increase in proliferation nor reduction in cell death, whereas other mutations may have profound effects on one or both functions. Consequently, attempting to calculate when pancreatic cancer began in a person by estimating cell numbers at diagnosis and cell-doubling time, and back-calculating to a hypothetical origin time, is without scientific basis in the context of the current understanding of the complexities of human cancer development.
Causes of Pancreatic Cancer
There are several host, genetic, and environmental causes of and associations with an increased risk of pancreatic cancer, and they are discussed elsewhere.3,6,7,27-30
The strongest association with pancreatic cancer is cigarette smoking, which is listed as a known human carcinogen by the U.S. National Toxicology Program (13th Report on Carcinogens), as carcinogenic to humans in general (class 1) by the International Agency for Research on Cancer (IARC), and specifically as a cause of pancreatic cancer by IARC. Other proposed associations and risk factors include age, race, geography, obesity, chronic pancreatitis, and diabetes. Risk factors less convincingly associated with pancreatic cancer include excess alcohol consumption (only in nonsmokers), diet (low fat and/or vegetarian), occupation, chemicals, pesticides, coffee consumption, gastrectomy, biliary disease including cholelithiasis, and ionizing radiation.
Several genetic disorders are associated with an increased risk of developing adenocarcinoma of the pancreas, including (1) hereditary breastand ovarian cancer syndrome caused by mutations in BRCA2; (2) familial melanoma caused by mutations in CDKN2A; (3) familial pancreatitis caused by mutations in PRSS1; (4) hereditary nonpolyposis colorectal cancer (Lynch syndrome), usually caused by a mutation in MLH1 or MSH2; (5) Peutz-Jeghers syndrome, caused by a mutation in STK11; and (6) von Hippel-Lindau syndrome, caused by mutations in VHL. There are also genetic disorders predisposing to cancers of the endocrine pancreas, including (1) neurofibromatosistype 1, caused by mutations in NF1; and (2) multiple endocrine neoplasia type 1, caused by mutations in MEN1.
There are also several forms of hereditary pancreatitis, some of which are associated with a markedly increased risk of pancreatic cancer. The best studied example is a syndrome of chronic pancreatitis with mutations in PRSS1. These persons have a 50-fold increased risk of developing pancreatic cancer compared with normal-risk individuals.31 Although these data support an association between chronic pancreatitis and pancreatic cancer, it is difficult to know whether the increased risk of pancreatic cancer results from the higher frequency of pancreatitis, a direct effect of the mutation, or both. And we always need to consider this may be an association and not cause and effect.
The relationship between sporadic chronic pancreatitis and pancreatic cancer is complex and controversial. Some meta-analyses and large case series report an increased relative risk estimate of pancreatic cancer in persons with chronic pancreatitis; others do not.6,7,32 Surveillance biases and confounding with alcohol consumption, cigarette smoking, and diet make definitive conclusions impossible. This association is further confounded by the observation that pancreatic cancer can cause and/or contribute to developing pancreatitis. A reasonable synthesis of current knowledge from a U.S. National Cancer Institute meeting seems appropriate: “Epidemiologically, pancreatitis can be considered as a disease continuum: starting with an attack of acute pancreatitis, leading to recurrent [acute pancreatitis] in some patients … progression to [chronic pancreatitis] in some … and ultimately of [pancreatic ductal adenocarcinoma].”27,29
The association between diabetes and pancreatic cancer is well established but is also complex (reviewed in references 33–36). Type 2 diabetes mellitus is associated with an increased risk of pancreatic cancer in many studies. This increased risk is greatest concurrent with a recent diagnosis of diabetes and declines as this interval increases. These data support the hypothesis that some cases of diabetes are a consequence of pancreatic cancer and are better termed type 3c diabetes. However, there remains an increased risk of pancreatic cancer more than 10 years after diabetes is diagnosed (reviewed in reference 37).
These data support the idea that diabetes may also cause and/or contribute to the development of pancreatic cancer. This remains to be proved. Because persons with diabetes often receive therapy, there may be confounding between increased or decreased insulin levels, other antidiabetic drugs, and pancreatic cancer risk. Some data suggest that insulin therapy may further increase pancreatic cancer risk, whereas other drugs such as metformin may decrease risk.38 Some other antidiabetes drugs have no discernible effect on pancreatic cancer risk, while others are controversial, requiring further study. A recent study suggested the association between diabetes and pancreatic cancer is indirect and mediated by a paraneoplastic syndrome,38 again emphasizing the distinction between an association and cause and effect.
There is a complex interplay among obesity, diabetes, acute and chronic pancreatitis, and pancreatic cancer. For example, obesity and diabetes are associated with insulin resistance. Obesity is also a risk factor for pancreatitis, which, in turn, is associated with an increased risk of pancreatic cancer. The sum of these data indicates that several reasonably clear exposures and conditions, regardless of mechanism, are associated with an increased risk of developing pancreatic cancer. Other associations are tenuous and/or complex.
In some instances, there is a convincing hereditary background for pancreatic cancer, mandating that we take a careful family history of cancers in everyone we see, not just for pancreatic cancer but for all cancers. This is often not carefully done. Some estimates suggest as many as 10% of cancers, especially those in young persons, may occur on a predisposing inherited genetic background.
Conclusion
Considerable progress has been made in our understanding of pancreatic cancer, but results of these advances are not yet evident in the clinic, either by way of earlier diagnosis or markedly better therapies. Pancreatic cancer is genetically complex, involving diverse intracellar pathways and having distinct mutational and transcriptome-defined subtypes, suggesting different pathogenic mechanisms. Will it be possible to use so-called precision oncology to tackle pancreatic cancer? I hope so. However, it is important to approach this problem with reasonable expectations.18,19
We are unlikely to manage pancreatic cancer with one or even several drugs in the way tyrosine kinase inhibitors have cured or operationally cured some people with CML. Different people are likely to need different therapies or combinations of therapies, which will need to be proved effective in controlled, ideally randomized clinical trials. And, as in all of medicine, prevention is better than therapy. Stop people from smoking, and we can probably decrease pancreatic cancer incidence by 10% to 20%. ■
Disclosure: Dr. Gale is a part-time employee of Celgene Corp.
References
4. Siegel R, Ma J, Zou Z, et al: Cancer statistics, 2014. CA Cancer J Clin 64:9-29, 2014.
5. Yeo TP: Demographics and epidemiology of pancreas cancer. Cancer J 18:477-484, 2012.
19. Tannock IF, Hickman JA: Limits to personalized cancer medicine. N Engl J Med 13:1289-1294, 2016.
34. Cui Y, Andersen DK: Diabetes and pancreatic cancer. Endocr Relat Cancer 19:F9-F26, 2012.
35. Li D: Diabetes and pancreatic cancer. Mol Carcinog 51:64-74, 2012.
36. Muniraj T, Chari ST: Diabetes and pancreatic cancer. Minerva Gastroenterol Dietol 58:331-345, 2012.