
Question 1: In this case, what is the most appropriate next best test?
Correct Answer: B. Peripheral blood smear examination.
Expert Perspective
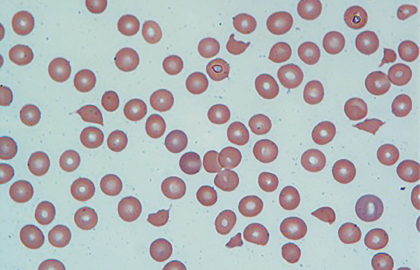
In the appropriate clinical setting, information obtained from a carefully examined peripheral blood smear film is indispensable. The peripheral blood smear, shown in Fig. 1, reveals schistocytes (“fragmented red blood cells” noted by arrows) consistent with microangiopathic hemolytic anemia and confirms “true” thrombocytopenia. These findings on peripheral blood smear can be observed in variety of conditions.1,2 Therefore, it is equally important to interpret them in the context of the patient’s clinical condition.
This clinical exercise will further narrow the differential diagnosis, which includes thrombotic thrombocytopenic purpura; Shiga toxin–associated hemolytic uremic syndrome (StxHUS or “typical” hemolytic uremic syndrome); atypical hemolytic uremic syndrome; disseminated intravascular coagulation; pregnancy-associated conditions such as preeclampsia, eclampsia, and HELLP syndrome; and severe “malignant” hypertension.
Although fragmented red blood cells with thrombocytopenia may be observed in the setting of severe cyanocobalamin deficiency,3,4 a normal white blood cell count, mean corpuscular volume, and a high reticulocyte count do not favor this diagnosis, and measurement of cyanocobalamin levels will not be useful. Ferritin measurement is not useful at this time, as current findings are not consistent with iron-deficiency anemia.
Question 2: Based on the patient’s clinical presentation and the peripheral blood smear film, what is the most likely diagnosis?
Correct Answer: C. Thrombotic thrombocytopenic purpura or hemolytic uremic syndrome.
Expert Perspective
It is late to expect preeclampsia, eclampsia, or development of HELLP syndrome in this postpartum patient, as she had no signs of these disorders prior to delivery except mild worsening of hypertension without proteinuria. Patients with HELLP syndrome develop right upper quadrant pain, transaminitis, microangiopathic hemolytic anemia, and low platelet count prior to delivery.
Although disseminated intravascular coagulation from retained products of conception, endometritis, or other infection is possible, the fibrinogen level is significantly higher than one would expect in the setting of acute disseminated intravascular coagulation.
Catastrophic antiphospholipid antibody syndrome and connective-tissue disorders (especially scleroderma renal crisis) may cause microangiopathic hemolytic anemia and thrombocytopenia.1,2,5 However, our patient’s clinical history and laboratory data do not support these diagnoses.
Our patient’s clinical presentation and laboratory and peripheral blood smear findings are most consistent with the diagnosis of thrombotic thrombocytopenic purpura or hemolytic uremic syndrome. Distinguishing between thrombotic thrombocytopenic purpura (acquired or congenital6) and hemolytic uremic syndrome (typical or atypical7,8) based on presenting clinical findings and initial laboratory tests is difficult, as there is a significant overlap in clinical symptoms and laboratory findings. Patients with thrombotic thrombocytopenic purpura often do not have the classic pentad of clinical findings that include microangiopathic hemolytic anemia, thrombocytopenia, fever, neurologic symptoms, and renal dysfunction, whereas 30% of patients with a diagnosis of hemolytic uremic syndrome will have fever and neurologic symptoms not commonly considered for the diagnosis of hemolytic uremic syndrome.1,2,5
Question 3: What test would aid in making the correct diagnosis prior to treatment?
Correct Answer: C. ADAMTS13 analysis.
Expert Perspective
ADAMTS13 is a metalloproteinase present in the blood responsible for cleaving ultralarge von Willebrand factor multimers into smaller-sized multimers. The ultralarge multimers are prothrombotic and can lead to the development of microvascular thrombosis and end-organ damage.
Deficiency in ADAMTS13 results in thrombotic thrombocytopenic purpura and can be acquired or congenital. Most cases of adult thrombotic thrombocytopenic purpura are acquired, due to the formation of immunoglobulin G autoantibody, which binds ADAMTS13 and clears it from the circulation. Congenital cases may be due to mutations resulting in low plasma levels of this protein, as in Upshaw-Schulman syndrome, which can manifest as thrombotic thrombocytopenic purpura at different ages.9-11
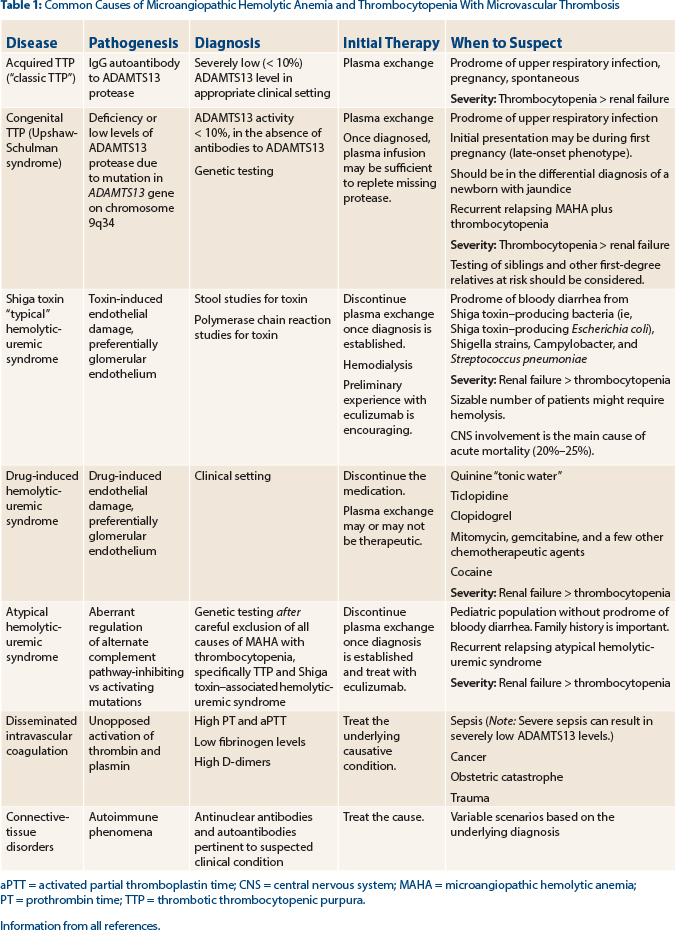
Tests for ADAMTS13 (activity levels and antibody detection) and Shiga toxin (produced by a variety of bacteria [see Table 1]) should be sent to support the diagnosis of thrombotic thrombocytopenic purpura vs Shiga toxin–associated hemolytic uremic syndrome, respectively. In our patient, the ADAMTS13 level was reported as less than 10% of normal, strongly supporting the diagnosis of thrombotic thrombocytopenic purpura.
Activity levels less than 10% are considered severely low and will confirm the diagnosis of thrombotic thrombocytopenic purpura in this patient. Activity levels in the normal range (greater than 50% in most laboratories) exclude the diagnosis of thrombotic thrombocytopenic purpura, despite the presence of microangiopathic hemolytic anemia, thrombocytopenia, and a normal coagulation profile. Activity levels between 10% and 50% are concerning but cannot be relied upon to make the diagnosis of thrombotic thrombocytopenic purpura with certainty, as decreases of these levels can be observed in a variety of inflammatory conditions.
Hemolytic uremic syndrome also results in the clinical and laboratory findings of microangiopathic hemolytic anemia and thrombocytopenia, but often the renal failure is a more dominant feature than the other “pentad” findings seen in thrombotic thrombocytopenic purpura. Hemolytic uremic syndrome can be classified as either typical or atypical, depending on the etiology.12
The most common form of hemolytic uremic syndrome occurs following infection (“typical hemolytic uremic syndrome”) with a Shiga-like toxin–producing bacteria with a prodrome of bloody diarrhea symptoms, as our patient experienced, but the tests were negative for Shiga toxin. Shiga-like toxin–producing Escherichia coli (STEC) strain O157:H7 is the most common pathogen resulting in typical STEC hemolytic uremic syndrome, but other bacteria, including Shigella strains, Campylobacter, and less frequently Streptococcus pneumoniae, can result in the development of typical hemolytic uremic syndrome.
In our patient, stool culture for organisms and Shiga toxin antigen detection assays (usually ELISA [enzyme-linked immunosorbent assay]–type assays) were negative. Polymerase chain reaction tests can be used to detect the presence of Shiga-toxin (Stx1 and Stx2) genes but are not widely available.12
Atypical hemolytic uremic syndrome is due to inherited mutations in the genes of complement regulatory proteins, resulting in excessive activation of the complement system. At least six mutated genes (CFH, CFI, MCP, CD46, C3, CFB, and THBD) and anti-CFH antibodies that can result in atypical hemolytic uremic syndrome have been identified,7,8,13 with the most common being CFH.
Measurement of complement levels in the setting of acute manifestations, however, is not useful in establishing the diagnosis, as levels can be low in a variety of acute and severe illnesses. Evaluation of genetic mutations is laborious, can take many months to report, and is costly. The tests can be done once common causes of microangiopathic hemolytic anemia and thrombocytopenia are excluded and are most useful in patients with relapsing courses of the disease. Although a von Willebrand factor multimer gel analysis may be abnormal in thrombotic thrombocytopenic purpura, results cannot be relied on to confirm the diagnosis of thrombotic thrombocytopenic purpura. ■
Disclosure: Drs. Abutalib and Connors reported no potential conflicts of interest.
References
1. George JN, Nester CM: Syndromes of thrombotic microangiopathy. N Engl J Med 371:654-666, 2014.
2. Tsai HM: Advances in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. J Am Soc Nephrol 14:1072-1081, 2003.
3. Tadakamalla AK, Talluri SK, Besur S: Pseudo-thrombotic thrombocytopenic purpura: A rare presentation of pernicious anemia. N Am J Med Sci 3:472-474, 2011.
4. Dalsania CJ, Khemka V, Shum M, et al: A sheep in wolf’s clothing. Am J Med 121:107-109, 2008.
5. Sadler JE: Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 112:11-18, 2008.
6. Levy GG, Nichols WC, Lian EC, et al: Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 413:488-494, 2001.
7. Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al: Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554-562, 2013.
8. Franchini M: Atypical hemolytic uremic syndrome: From diagnosis to treatment. Clin Chem Lab Med. March 18, 2015 (early release online).
9. Furlan M, Robles R, Lämmle B: Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood 87:4223-4234, 1996.
10. Tsai HM: Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood 87:4235-4244, 1996.
11. Moatti-Cohen M, Garrec C, Wolf M, et al: Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. French Reference Center for Thrombotic Microangiopathies. Blood 119:5888-5897, 2012.
12. Trachtman H, Austin C, Lewinski M, Stahl RA: Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol 8:658-669, 2012.
13. Gruppo RA, Rother RP: Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med 360:544-546, 2009.