
Question 1: What is the next best step for this patient?
Correct Answer: C. Determination of amyloid subtype.
Expert Perspective
Amyloidosis encompasses a heterogeneous group of diseases bound by the characteristic deposition of amyloid fibrils in soft tissues and bone marrow, and it could be either systemic or localized. The most frequent categories are light chain (AL); derived from monoclonal B-cell disorders, which produce the amyloidogenic protein; hereditary; and senile systemic (ATTR; transthyretin amyloidosis) variants from mutant and wild-type transthyretin (TTR).1,2
A review of systemic amyloidoses occurring in humans is shown in Table 1.3 Timely and accurate differentiation between these amyloid subtypes cannot be overemphasized; the clinical exercise of classifying amyloidosis is prudent, since management varies dramatically between different subtypes. Therefore, upon declaration of a positive Congo red stain, further testing to classify amyloid protein must be triggered immediately (choice C). The concomitant presence of serum and/or urine monoclonal proteins does not always imply a diagnosis of AL amyloid.1,2 For example, nearly 25% of patients with endomyocardial biopsy–proven ATTR (non-AL amyloid) can have concurrent monoclonal gammopathy of undetermined significance.4
Plasma cell–directed therapy (choice A) is not justified without further testing. Emergent cardiac catheterization (choice B) would be a reasonable choice in the case of ischemic cardiac disease, which is absent in our patient.
Question 2: Which statement about this patient’s condition is correct?
Correct Answer: A. Localized amyloidosis should be considered in the differential diagnosis.
Expert Perspective
Diabetic patients on subcutaneous insulin injections can develop “localized” (injection site, which is mostly abdomen) insulin amyloidomas.5 The presence of diabetic complications (ie, peripheral neuropathy, proteinuria, diastolic dysfunction, along with a positive Congo red stain on SFA) definitively carries a risk of misdiagnosis. There have been reported cases of monoclonal gammopathy of undetermined significance with positive Congo red deposits on SFA in patients who were later diagnosed with iatrogenic “localized” insulin amyloidosis and not systemic amyloidosis.6 Therefore, following a positive Congo red stain, the next best step is to perform amyloid subtyping on positive SFA (choice C).7-9
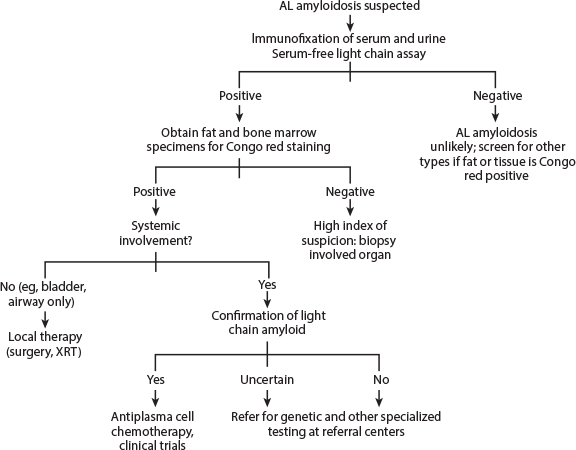
Fig. 1: Diagnostic approach to AL amyloidosis. AL = amyloid light chain; XRT = radiotherapy. From Merlini G, et al: J Clin Oncol 29:1924-1933, 2011.
In this patient with infiltrative cardiomyopathy, neuropathy, positive Congo red stain, and monoclonal gammopathy of undetermined significance, the original clinical impression is AL amyloidosis, but caution is advised! Both localized and systemic amyloidosis must be considered, but again, accurate subtyping of the amyloid deposits is a prerequisite to the correct management of this heterogeneous disease.
Question 3: Which statement about mass spectrometry–based proteomic analysis is correct?
Answer: A. Mass spectrometry–based proteomic analysis can classify amyloidosis.
Expert Perspective
The rationale of proteomics in amyloid typing stems from the presence of large numbers of peptides in the amyloidogenic protein, concurrent with apolipoprotein E and serum amyloid P component. Mass spectrometry–based proteomic analysis is the gold standard test, with its applicability even on a limited tissue sample ([60,000 µ2]7-9; choice B). Furthermore, this technique can help identify new proteins and variants that can cause amyloid deposition disease.
Mass spectrometry–based proteomic analysis for screening amyloidosis can be applied with high sensitivity and specificity on positive SFA9 (choice C). Alternatively, immunohistochemistry method may be used. However, this test lacks reliability and sensitivity compared with mass spectrometry–based proteomics. In patients with known amyloidosis, as many as 65% of Congo red–negative SFA can show the classic amyloid signature, suggesting the role of mass spectrometry–based proteomic analysis, even in SFA-negative samples in patients with a high clinical suspicion of amyloidosis.9 At this time, however, in clinical practice, if an SFA and bone marrow biopsy are negative for Congo red stain and the clinical suspicion for amyloidosis remains high, biopsy of the involved organ is recommended. Fig. 1 highlights the diagnostic approach to AL-amyloidosis.10
Question 4: If this patient has non-AL amyloid, what is the best management of underlying plasma cell disorder?
Correct Answer: B. Observation.
Expert Perspective
In the setting of non-AL amyloid, the underlying plasma cell disorder would be best classified as monoclonal gammopathy of undetermined significance (ie, M spike < 3 g/dL, < 10% clonal plasma cells in bone marrow, and absence of end-organ damage attributable to the plasma cell disorder).11 Risk stratification of monoclonal gammopathy of undetermined significance11 in this patient allocates him to low-intermediate risk, which implies he has a 21% risk of progression to malignancy at 20 years. Patients with monoclonal gammopathy of undetermined significance are at higher risk than the general population for the development of osteoporosis/osteopenia, peripheral neuropathy, and thromboembolic events.11
Expert consensus recommendations include assessment of bone mineral density by dual-energy x-ray absorptiometry scan at diagnosis.11 For patients with intermediate-risk monoclonal gammopathy of undetermined significance, the International Myeloma Working Group recommends follow-up with monoclonal protein studies in 6 months and then annually for life.11 No treatment is indicated unless it is part of a clinical trial. Repeat bone marrow biopsy is not recommended if serum and urine monoclonal proteins remain stable (choice C).
Concluding Remarks
In this patient, based on the kappa light chain elevation and presence of plasma cell disorder, the initial clinical impression was AL amyloidosis. The Congo red deposits in the SFA were subject to mass spectrometry–based proteomic analysis and determined to be ATTR. Transthyretin DNA-sequencing studies did not reveal mutations, which was consistent with wild-type ATTR or age-related amyloidosis (previously referred to as senile systemic amyloidosis). This is an acquired disorder mainly affecting men over the age of 60 years, resulting from the deposition of wild-type TTR amyloid.12
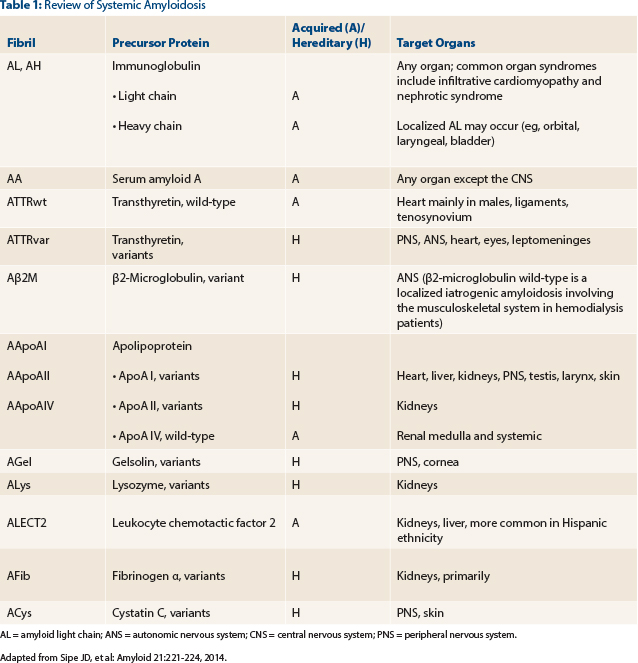
AL = amyloid light chain; ANS = autonomic nervous system; CNS = central nervous system; PNS = peripheral nervous system. Adapted from Sipe JD, et al: Amyloid 21:221-224, 2014.
Although a regimen of bortezomib (Velcade), cyclophosphamide, and dexamethasone followed by autologous hematopoietic cell transplant was initially considered for this patient with ATTR amyloid, this approach would be harmful and ineffective. The current recommended treatment for age-related ATTR is largely supportive; however, tafamidis (an oral selective stabilizer of TTR) has been shown to stabilize the TTR tetramer by binding with very high affinity to the two thyroxine-binding sites, preventing the tetramer from dissociating into the amyloidogenic monomers.13 The randomized controlled phase III clinical trial evaluating the use of tafamidis (NCT01994889) completed enrollment in August 2015 and will provide answers regarding its utility in wild-type ATTR. Doxycycline with tauroursodeoxycholic acid (NCT01855360) and the nonsteroidal anti-inflammatory agent diflunisal may have a role in managing age-related ATTR based on the results of few small studies.14 ■
Disclosure: Drs. Abutalib and D’Souza reported no potential conflicts of interest.
References