

What is different about our trials vs other CAR T trials is that we believe the composition of the T cells is critically important…. Our study is the first to give an equal mix of CD4-positive and CD8-positive cells, and we hope this gets us a better therapeutic index.— David G. Maloney, MD, PhD
Tweet this quote
Research conducted at the Fred Hutchinson Cancer Research Center, Seattle, is moving the field of chimeric antigen receptor (CAR) T-cell therapy forward in the treatment of B-cell malignancies. At the 2016 Pan Pacific Lymphoma Conference in Koloa, Hawaii, David G. Maloney, MD, PhD, Professor of Medicine at the University of Washington and Member of the Clinical Research Division, Fred Hutchinson Cancer Research Center, described promising results in patients with non-Hodgkin lymphoma who received his group’s own CAR T-cell construct.1
The product engineered at Fred Hutchinson is a second-generation CAR T-cell product that comprises a murine FMC-63 scFV (single-chain fragments of the antibody variable region) antibody against CD19, with a 4-1BB costimulatory domain (some other CAR T cells incorporate C28 as a costimulatory domain; first-generation CAR T cells lacked costimulatory domains). The CAR T cells are marked by coexpression of a truncated epidermal growth factor receptor (EGFRt). The Fred Hutch CD-19 CAR T cells are distinct in that they are a mix of CD8-positive and CD4-positive–derived CAR T cells, which provided optimal potency in a preclinical model, he explained.
“We found that not all T cells are the same. What is different about our trials vs other CAR T trials is that we believe the composition of the T cells is critically important, so we selected CD4-positive and CD8-positive T cells independently and used an equal mix. This composition has potent antitumor activity,” revealed Dr. Maloney. “Our study is the first to give an equal mix of CD4-positive and CD8-positive cells, and we hope this gets us a better therapeutic index.”
Dr. Maloney emphasized that CAR T cells are “a living drug” that requires a small amount of product to produce a robust effect. “You can give an incredibly small amount, less than 20 million total cells, which is a tiny pellet in the bottom of a test tube, and within 3 to 4 days, you have 10 times more than you started with,” he continued. “By day 8 after infusion, up to 90% of cells in the blood can be CAR T cells that are expanding and going after the tumor.”
Protocol 2639
Dr. Maloney and his team have treated 125 patients with B-cell malignancies on Protocol 2639 (41 of whom had non-Hodgkin lymphoma [NHL]). The study is examining the feasibility of manufacturing a CD19 CAR T-cell product of defined composition and is determining the safest dose. Unlike some other trials, their study does not exclude patients on the basis of absolute lymphocyte count, circulating tumor, or prior transplant, or require a test of T-cell expansion.
Phase I of the study tested three dose levels of CAR T cells manufactured from bulk CD4-positive T cells and CD8-positive cells: 2 × 105/kg, 2 × 106/kg, and 2 × 107/kg. The first 12 patients received cyclophosphamide with or without etoposide for lymphodepletion.
The investigators were surprised to detect anti-CAR immune responses in some of these patients, as evidenced by a loss of CD19 CAR T cells and B-cell recovery within 100 days of infusion.
“We found we had a robust expansion of CAR T cells in the majority of patients, but their CAR T-cell persistence was quite low and abruptly vanished before day 28. The problem was that patients were making a T-cell–mediated immune response against the CAR T-cell construct, and this was causing rejection of the CAR T cells,” Dr. Maloney said.
They hypothesized that intensification of the lymphodepletion could improve outcomes. They modified the regimen to include cyclophosphamide and fludarabine, which led to higher expansion of the CAR T cells and to greater persistence. The regimen reduces transgene immune responses and enables responses to a second CAR T-cell infusion, he revealed.
Although dose level 3 had previously been associated with no dose-limiting toxicities, the cyclophosphamide/fludarabine regimen did demonstrate more toxicity. Dose level 2 with this lymphodepletion regimen was deemed most acceptable, he added.
Responses Highest With Modified Regimen
Dr. Maloney reported the results for the first 41 patients with NHL, of whom 73% had aggressive subtypes and 66% had received at least four prior lines of therapy. The CAR T-cell product was successfully manufactured for all the patients, and 68% of the patients were able to receive infusions as outpatients.
Of the 41 patients, 39 were evaluable; 26 (67%) responded, and 13 (33%) were complete responses. Response rates were further improved to 80% with cyclophosphamide/fludarabine lymphodepletion at dose level 2 (Table 1).
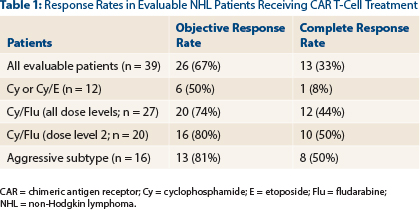
“The addition of fludarabine was associated with an increasing quality of response (P = .02),” he noted. “Responses were seen in multiple histologies.”
Factors associated with a significantly increased likelihood of achieving a response were higher levels of CD4-positive and CD8-positive EGFRt-marked cells and lymphodepletion with cyclophosphamide/fludarabine vs cyclophosphamide alone or with etoposide.
Dr. Maloney emphasized the importance of achieving a complete response. For the cyclophosphamide/fludarabine lymphodepletion group who achieved a complete response to treatment at dose level 2 (the optimal dose), median progression-free survival has not been reached, whereas it was 4.1 months in patients with less-robust responses. All these patients are alive at a median follow-up of 7 months, compared with approximately 70% of those who did not achieve a complete response.
Dose-Toxicity Relationship
The researchers also observed a relationship between the infused CAR T-cell dose and toxicity. Toxicity was predominantly cytokine-release syndrome, which occurred in 63% and was severe in four patients (13%), two of whom died at the highest dose level (2 × 107/kg) and neurotoxicity (mainly encephalopathy), which was grade 3 to 4 in 25% of the patients.
Cytokine-release syndrome and neurotoxicity both seemed to be more likely in acute lymphocytic leukemia patients compared with non-Hodgkin lymphoma patients.
CAR T Cells for Lymphoma
- Researchers at the Fred Hutchinson Cancer Research Center have engineered a CAR T-cell product that is an equal mix of CD8-positive and CD4-positive–derived CAR T cells; this composition has shown optimal potency in a preclinical model.
- Protocol 2639 has enrolled 125 patients with B-cell malignancies, and for 39 evaluable patients with non-Hodgkin lymphoma, the response rate to CAR T-cell therapy was 67%, with a 33% rate of complete responses.
- In patients who received the optimal lymphodepletion regimen (cyclophosphamide/fludarabine) at the optimal dose level, response rates improved to 80%, with 50% being complete responses.
Dr. Maloney and his colleagues, Cameron Turtle, MBBS, PhD, and Stanley R. Riddell, MD, are evaluating the levels of cytokines and other factors in patients who do and do not develop neurotoxicity and cytokine-release syndrome to determine whether a certain baseline profile might predict for these toxicities. So far, they have observed that higher levels of interleukin (IL)-6 and IL-15 and low levels of transforming growth factor (TGF)-beta on day 1 after CAR T-cell infusion are associated with ≥ grade 3 neurotoxicity.
“In the future, we may be able to give CAR T cells and do a cytokine assay to predict those patients for whom we need to consider measures to slow down the CAR T-cell growth,” he predicted.
“Our results are very encouraging for the field, but there’s still a lot to learn. We need to overcome the issue of immunogenicity to allow T cells to persist longer, and minimize toxicity,” he added. “Are we curing patients? It’s still hard to know.” ■
Disclosure: Dr. Maloney has received research funding from Juno Therapeutics.
Reference